

Vidhi Lalchand
Postdoctoral Fellow, Broad and MIT
Vidhi Lalchand*, Aditya Ravuri*, Emma Dann*, Natsuhiko Kumasaka, Dinithi Sumanaweera, Rik G.H. Lindeboom, Shaista Madad, Sarah A. Teichmann, Neil D. Lawrence
Machine Learning and Computational Biology , 2022
Single-cell RNA-seq datasets are growing in size and complexity, enabling the study of cellular composition in various biological and clinical contexts.
Dimension reduction techniques are an essential precursor to downstream tasks like cell type clustering, pseudotime estimation and sub-population identification.
Models that account for technical and biological confounders (e.g. batch effect, inter-individual variation, proliferation signatures) are required
Motivation
Single-cell RNA-seq datasets are growing in size and complexity, enabling the study of cellular composition in various biological and clinical contexts.
Dimension reduction techniques are an essential precursor to downstream tasks like cell type clustering, pseudotime estimation and sub-population identification.
Models that account for technical and biological confounders (e.g. batch effect, inter-individual variation, proliferation signatures) are required
Motivation
Gaussian Process Latent Variable Models (GPLVMs) for single-cell data
Trajectory analysis
(GrandPrix, Ahmed et al. 2019)
Exploratory analysis
(Verma and Engelhardt et al. 2022)
Spatio-temporal modelling
(MEFISTO, Velten et al. 2022)
Single-cell RNA-seq datasets are growing in size and complexity, enabling the study of cellular composition in various biological and clinical contexts.
Dimension reduction techniques are an essential precursor to downstream tasks like cell type clustering, pseudotime estimation and sub-population identification.
Models that account for technical and biological confounders (e.g. batch effect, inter-individual variation, proliferation signatures) are required
Motivation
Gaussian Process Latent Variable Models (GPLVMs) for single-cell data
Trajectory analysis
(GrandPrix, Ahmed et al. 2019)
Exploratory analysis
(Verma and Engelhardt et al. 2022)
Spatio-temporal modelling
(MEFISTO, Velten et al. 2022)
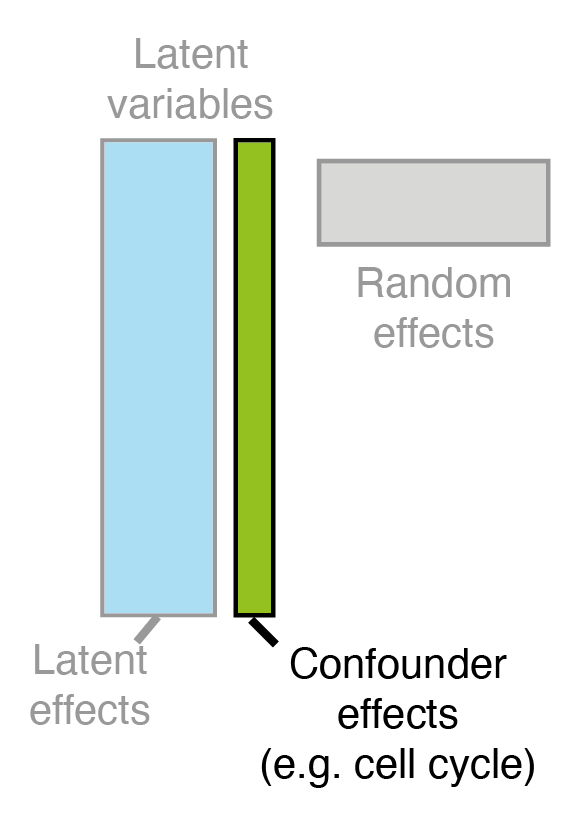
Limitations: bad scalability to large datasets, missing modelling of confounders
Given: High dimensional training data \( Y \equiv \{\bm{y}_{n}\}_{n=1}^{N}, Y \in \mathbb{R}^{N \times D}\)
Learn: Low dimensional latent space \( X \equiv \{\bm{x}_{n}\}_{n=1}^{N}, X \in \mathbb{R}^{N \times Q}\)
N x D
The mathematical bridge
2d latent space (each point represents a cell)
High-dimensional data space (A cell by gene matrix of expression counts)
A visualisation of the workings of a latent space model
Application: Understanding cellular similarities from a single-cell gene expression matrix
The Model: The Kernel function
The inductive biases of the GP mapping are controlled by a kernel function
N x D
D - independent Gaussian processes
low dimensional latent space (Q)
High-dimensional data space (D)
\( f_{d} \sim \mathcal{GP}(0, k_{f})\)
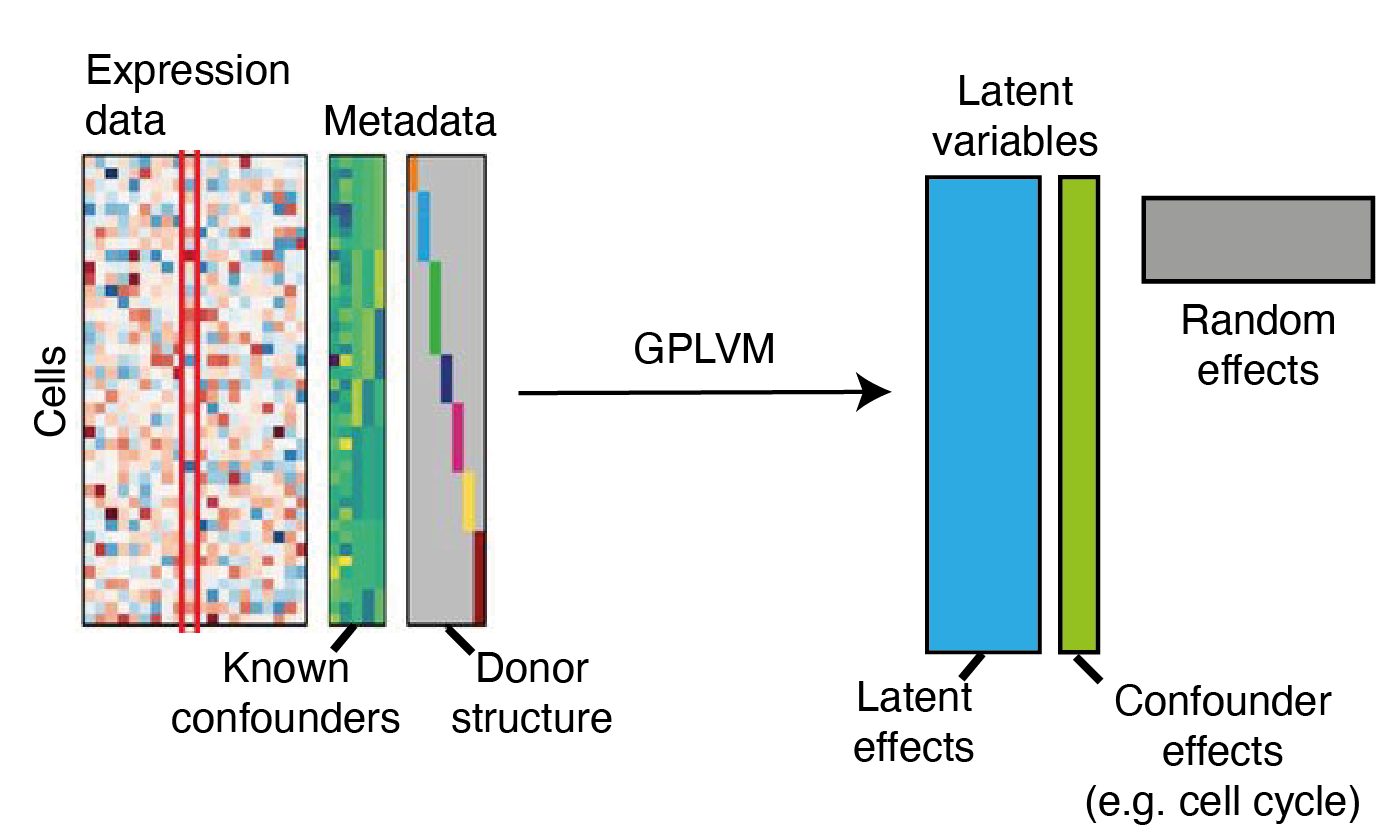
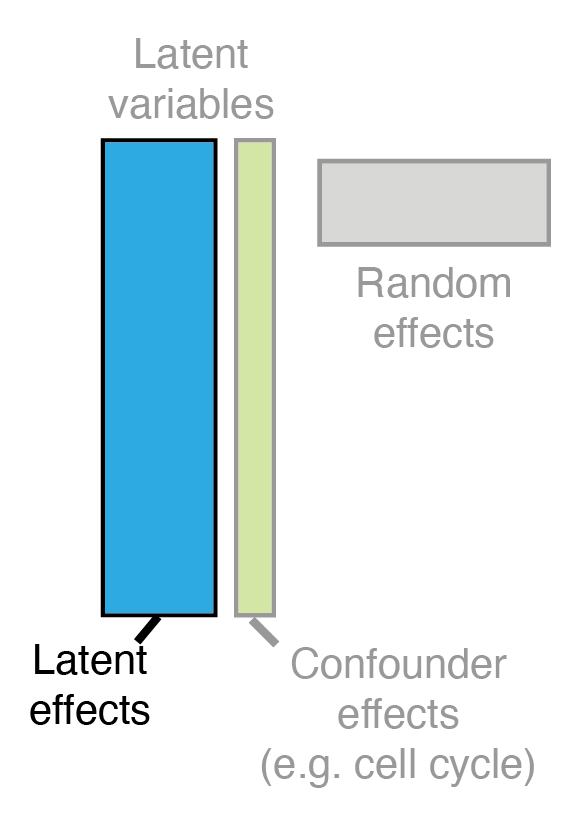
The Model: Augmented Kernel Function
where we assume a constant mean \(\mu_{f} \in \mathbb{R} \) for the \(\bm{f}\) process, the design matrix \(\Phi\) with covariates is specified and \(\zeta_{d}\) encapsulates the mean of random effects \(B\).
The expression matrix \(Y\) is driven by this joint process \(\tilde{F}\) with columns \(\tilde{f}_{d}\) distributed as individual Gaussian processes
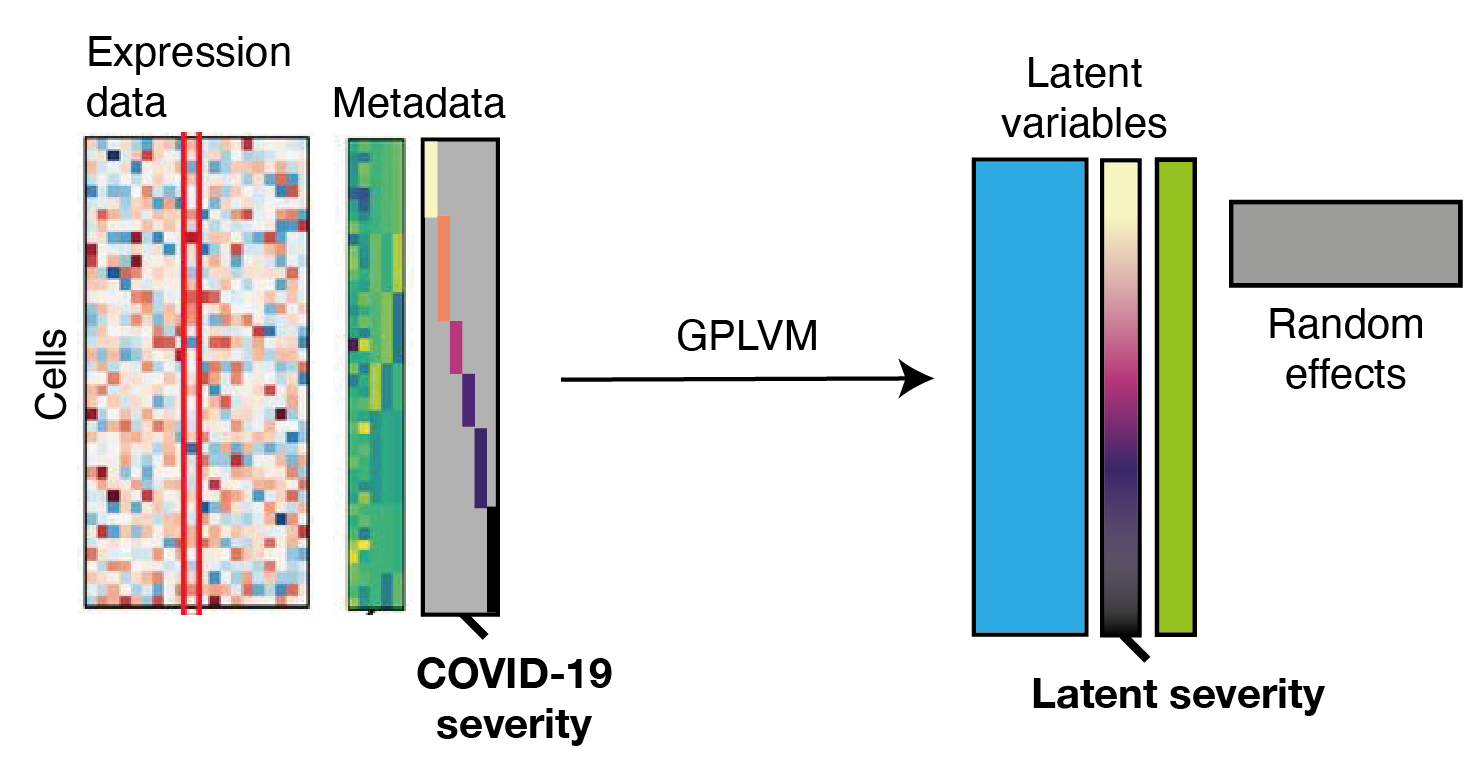
Metadata
Expression
data
The Model: Stochastic Variational Inference
Canonical GP prior
Augmented GP prior
The augmented kernel formulation allows us to derive an objective (evidence lower bound) which factorises across both cells (\(N\)) and genes (\( D\)).
*Details about the derivation of the objective are in the paper
While not converged do
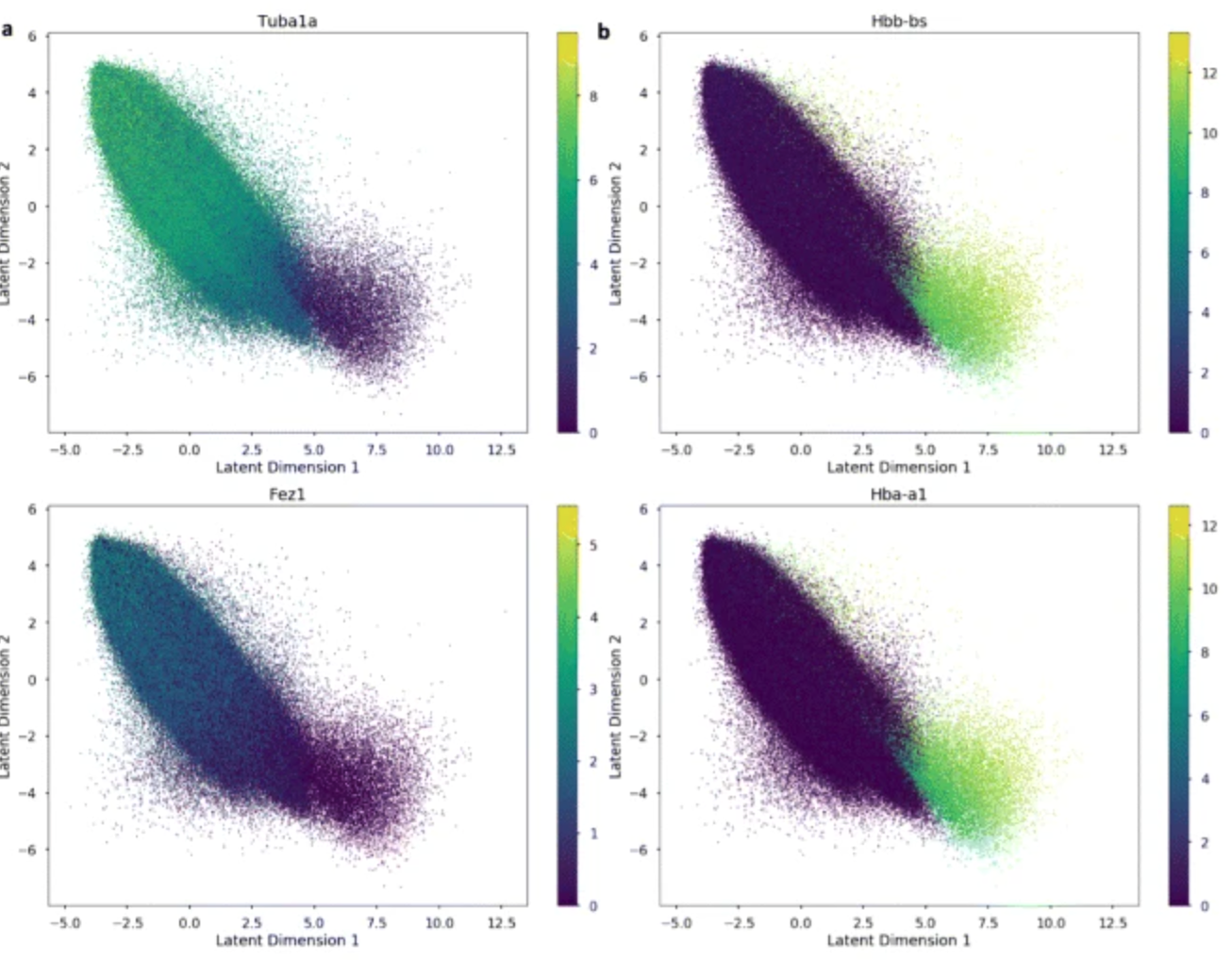
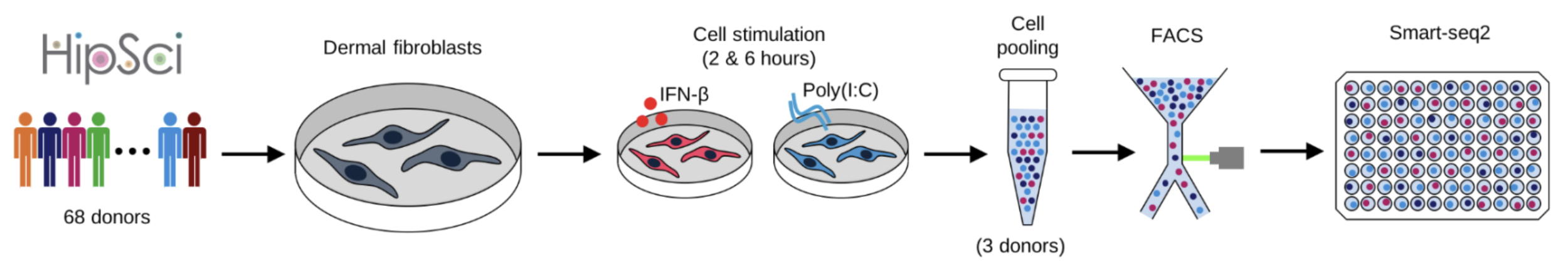
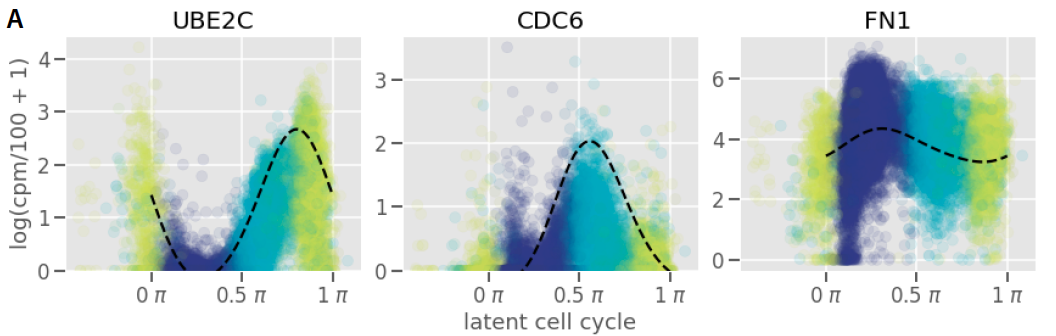
Application 1: Reproducing innate immunity analysis
Kumasaka et al. (2021) Mapping interindividual dynamics of innate immune response at single-cell resolution. biorXiv
Application 1: Reproducing Innate immunity analysis
Kumasaka et al. (2021) Mapping interindividual dynamics of innate immune response at single-cell resolution. biorXiv
Application 1: Reproducing Innate immunity analysis
Kumasaka et al. (2021) Mapping interindividual dynamics of innate immune response at single-cell resolution. biorXiv
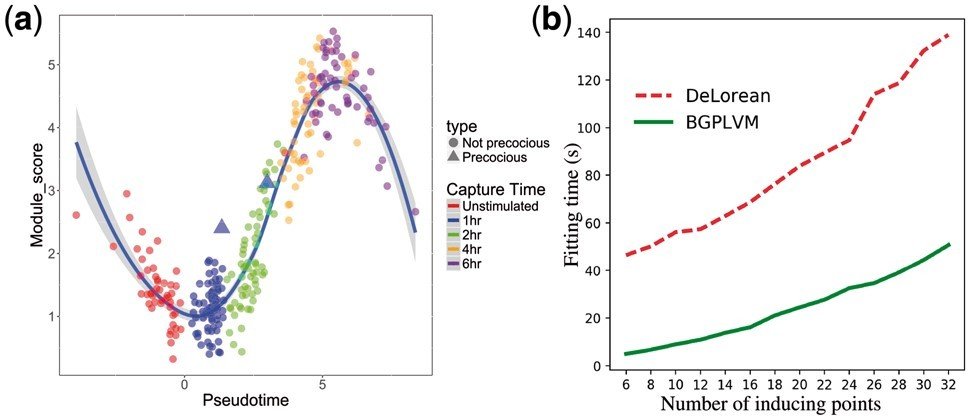
Runtime comparison
Additive GPLVM (Kumasaka et al.): 4.5 hrs
Augmented GPLVM: 30 mins
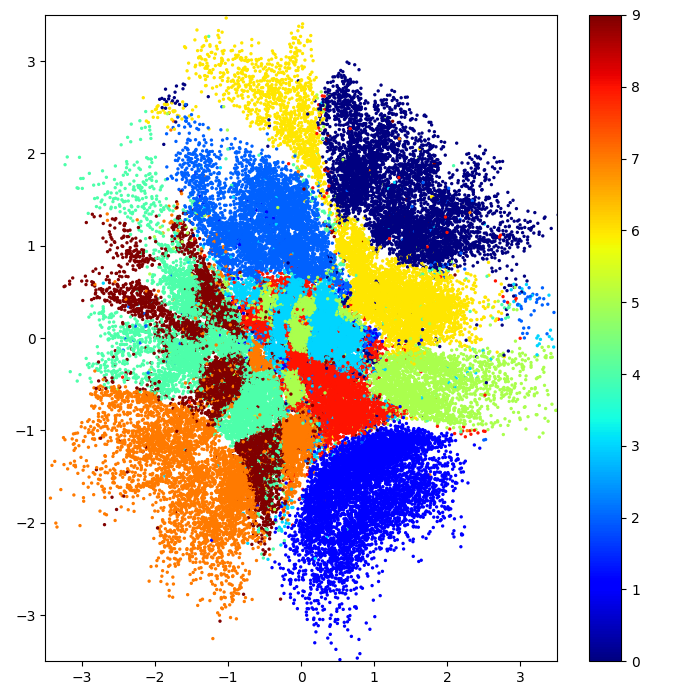
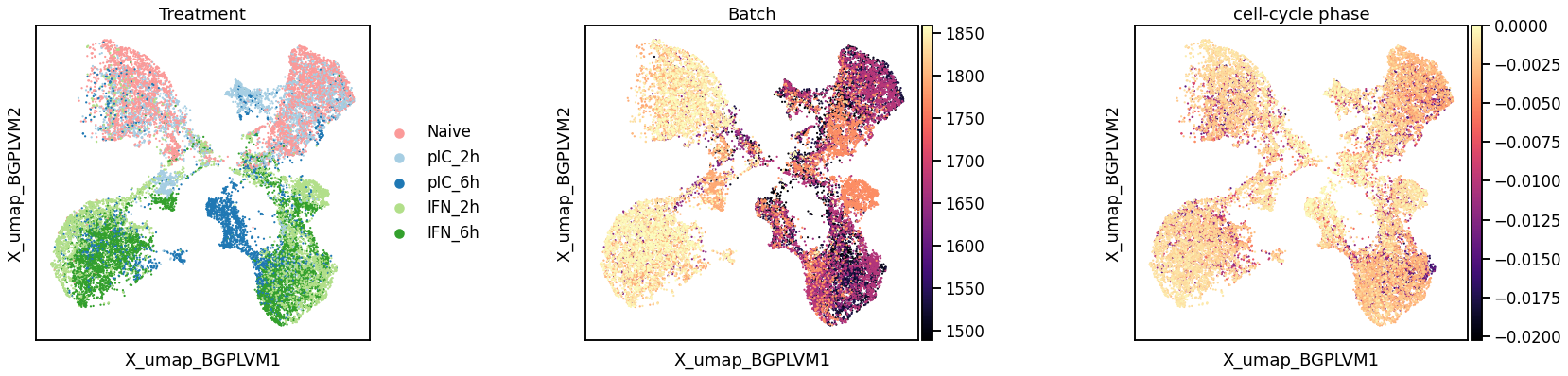
Augmented kernel disentangles cell cycle and treatment effects
G1 phase
S phase
G2 phase
Treatment
Latent batch effect
Cell cycle phase
Augmented kernel disentangles cell cycle and treatment effects
Application 2: COVID-19 scRNA-seq cohort
Data: Stephenson et al. (2021) Single-cell multi-omics analysis of the immune response in COVID-19. Nat Medicine
54,941 cells
130 patients
PCA
scVI
Augmented GPLVM
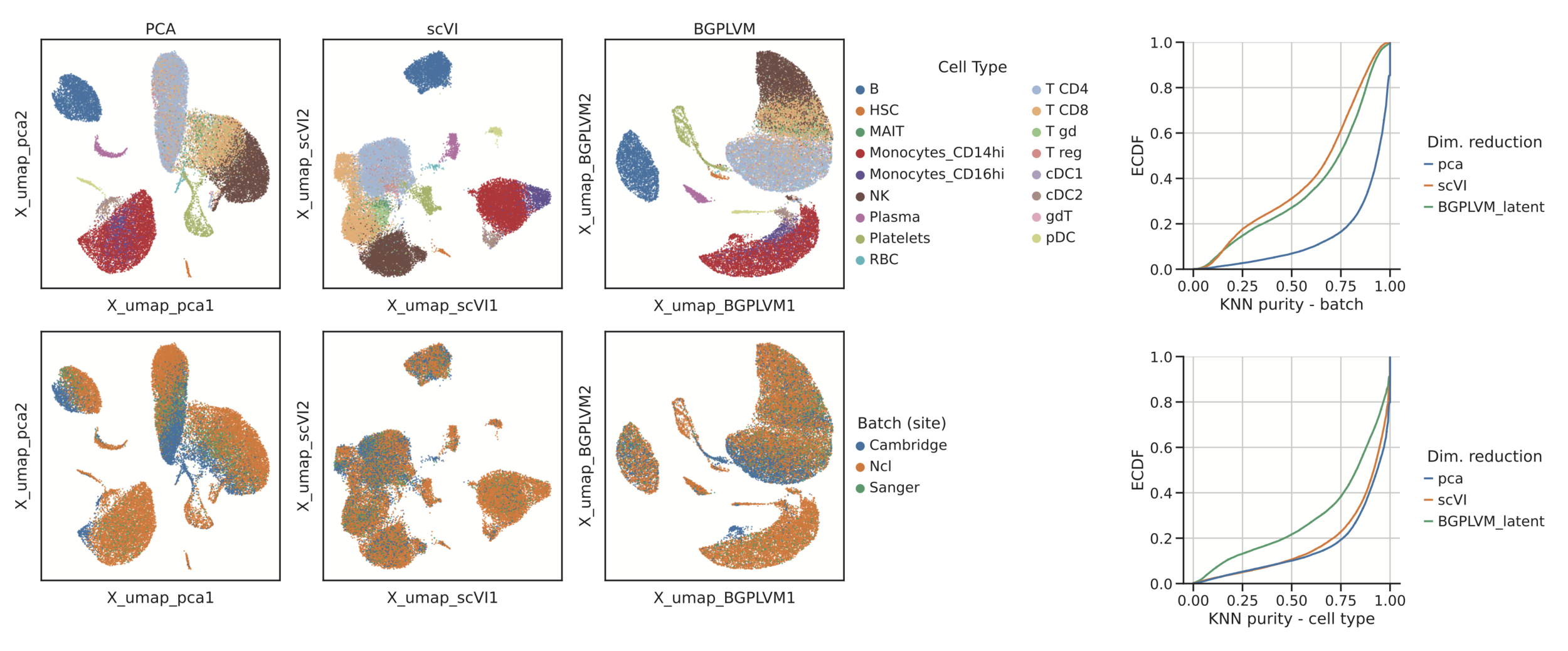
Application 2: COVID-19 scRNA-seq cohort
Data: Stephenson et al. (2021) Single-cell multi-omics analysis of the immune response in COVID-19. Nat Medicine
PCA
scVI
Augmented GPLVM
54,941 cells
130 patients
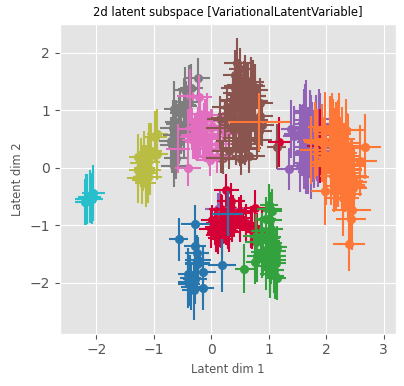
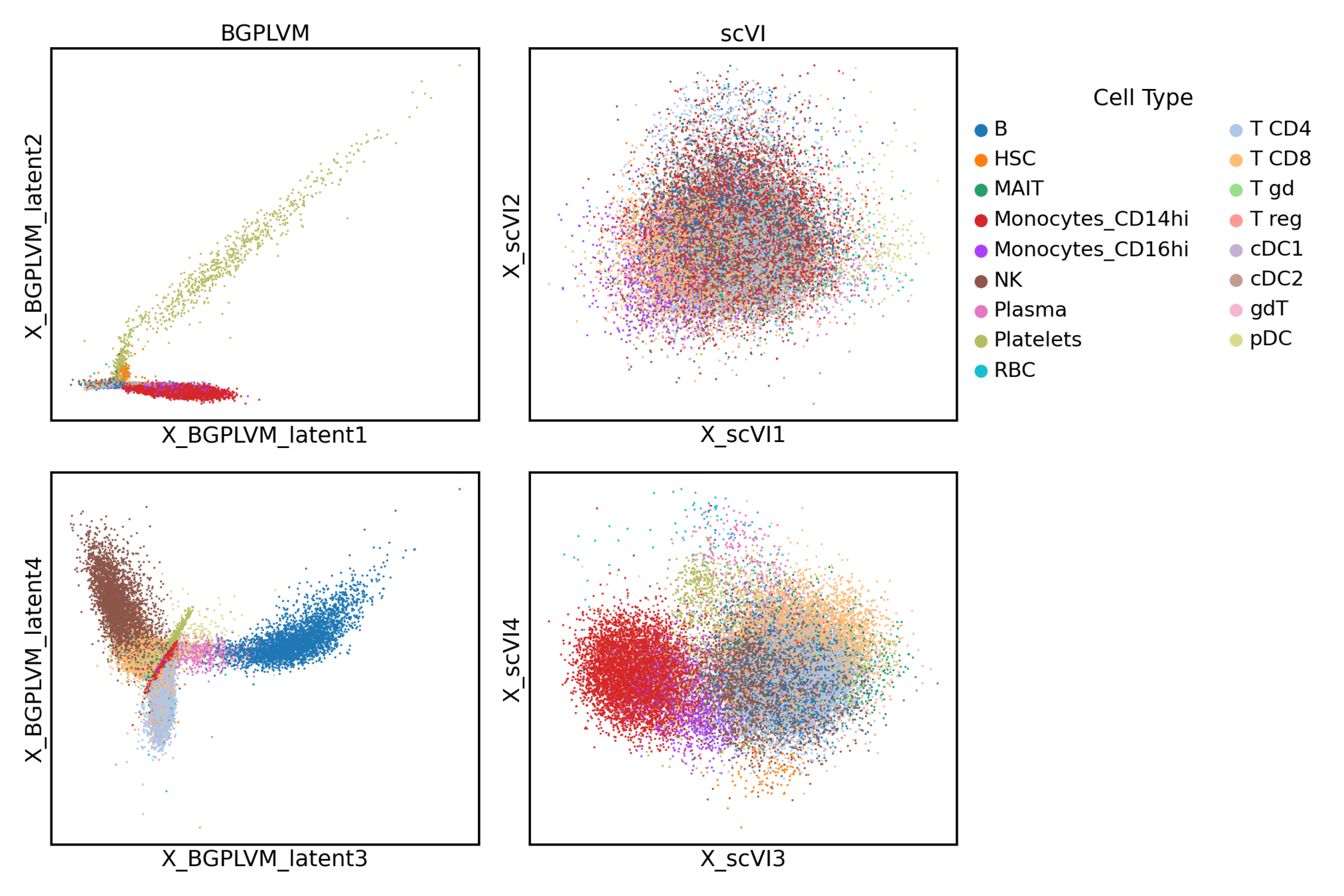
The augmented GPLVM learns interpretable latent dimensions
scVI
Augmented GPLVM
scVI
Augmented GPLVM
The augmented GPLVM learns interpretable latent dimensions
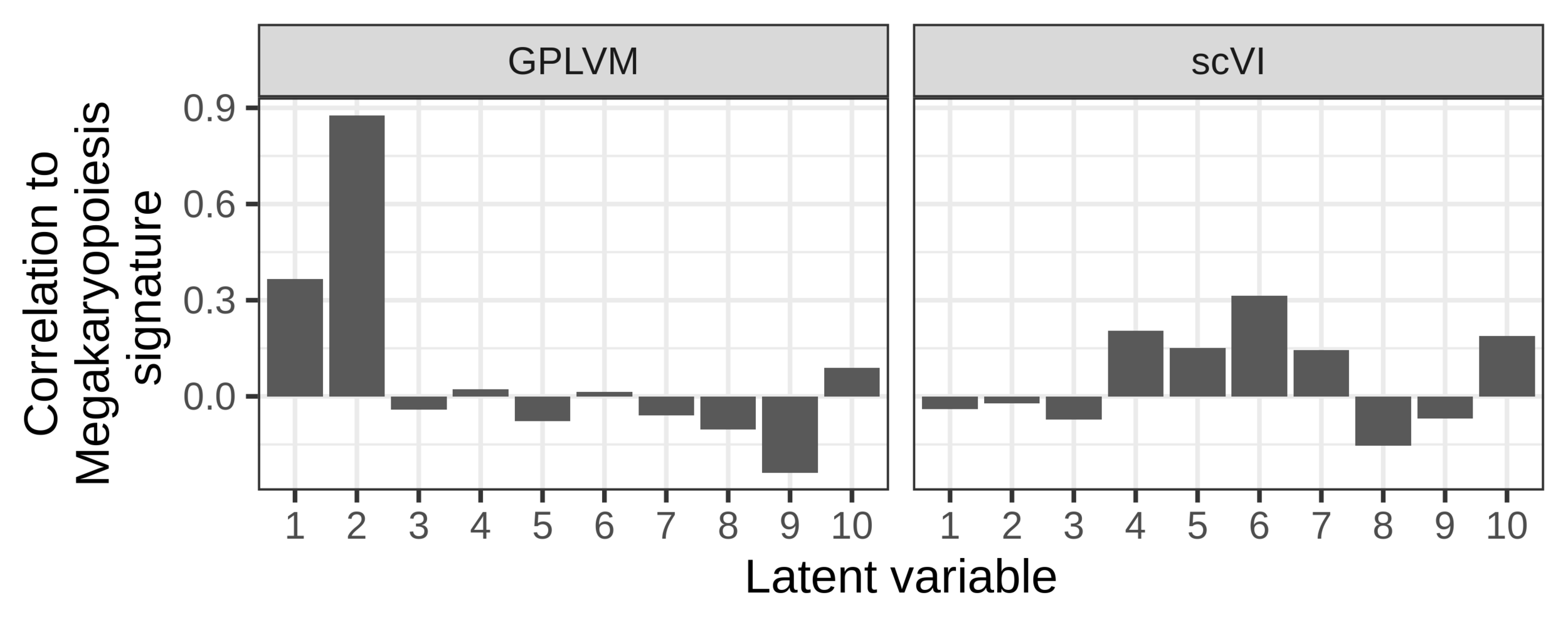
scVI
Augmented GPLVM
Correlation to platelet differentiation signature
The augmented GPLVM learns interpretable latent dimensions
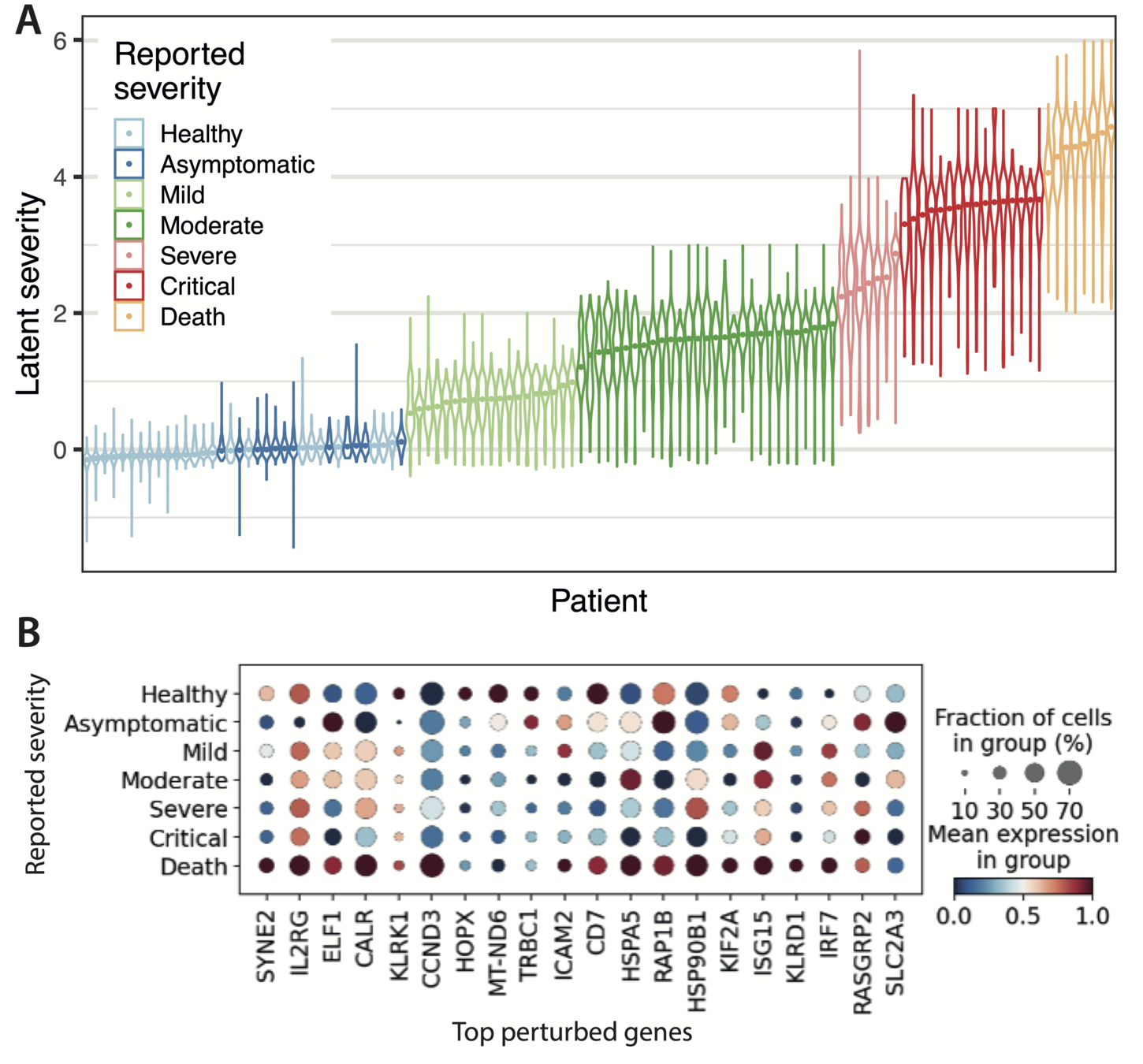
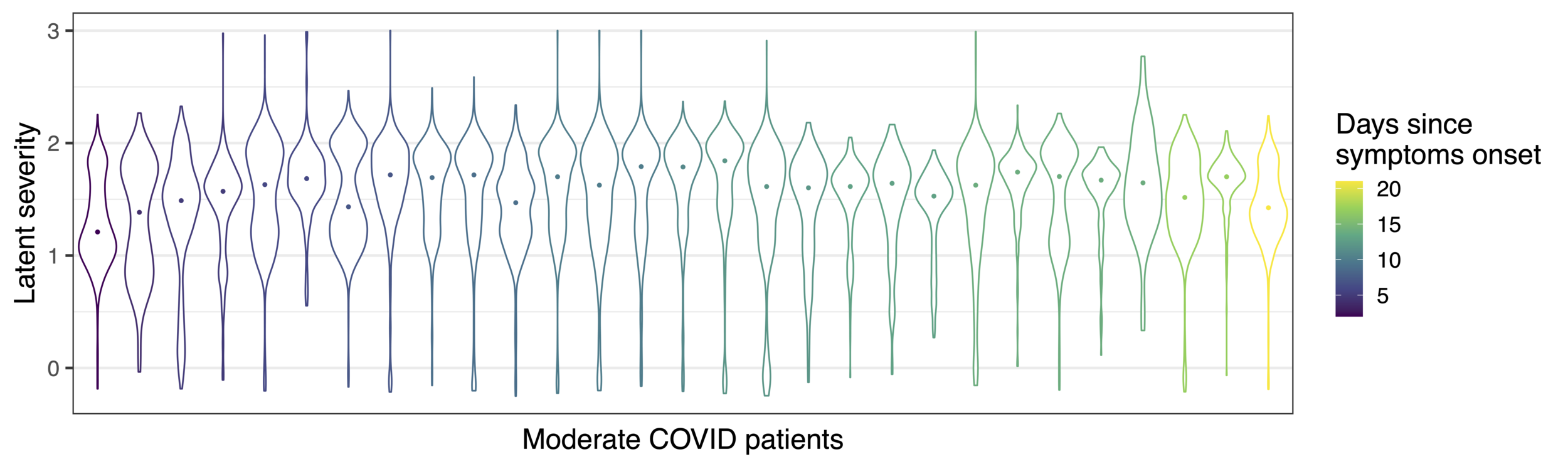
Modelling biological variation: COVID-19 severity
Modelling biological variation: COVID-19 severity
Latent severity captures variation in days since onset of symptoms
R2 = 0.25, p-val < 2e-16
GPLVM generative model recovers signatures of COVID-19 severity
Reported severity
Top perturbed genes
(highest variance upon perturbation)
GPLVM generative model recovers signatures of COVID-19 severity
Reported severity
Top perturbed genes
(highest variance upon perturbation)
Cell type markers
Viral entry factors
Interferon response
Summary
We introduce an augmented kernel function that jointly models known and unknown technical and biological covariates in scRNA-seq datasets
The formulation amenable to SVI enables application to complex cohort studies
Next steps:
Further scale-up with amortised inference
Modelling complex covariance structure (e.g. genotype effects for eQTL analysis)
Acknowledgements
Neil Lawrence
Vidhi Lachland
Adytia Ravuri
Sarah Teichmann
Emma Dann
Dinithi Sumanaweera
Rik Lindeboom
Shaista Madad
By Vidhi Lalchand



